








|
|||||||||||||||||||||
|
|
Supervisor : Dr. Thierry MARTENS Director : Pr. Jacques ROYER
Synthèse et Structure des Composés d' Interêt Pharmacologique (Website) UMR 8638, Faculté de Pharmacie 4, avenue de l'Observatoire, 75006 PARIS
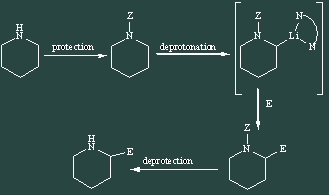
Functionalization a to a nitrogen is often a key step in the synthesis of natural products like alkaloids or amino-acids. Starting from a simple "naked" amine, this transformation can be achieved more commonly using two paths: an anionic and a cationic one. T he anionic path consists in the deprotonation in the a position using a strong base (BuLi) to generate an anionic intermediate that can react with various electrophiles. This methodology has been well developed, including a stereoselective version using either a chiral auxiliary or a chiral inductor.
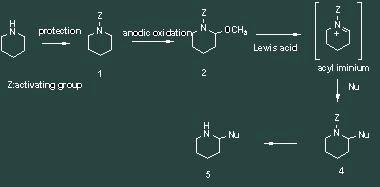
Scheme 1 he cationic path uses an anodic oxidation to activate the C-H bond a to the nitrogen. This sequence, known as Shono's sequence, starts with the protection of an amine with a suited deactivating group. In an electrochemical step, an iminium is intermediately generated by oxidation to be trapped by methanol, which is the solvent of the reaction. This methoxylated product is a stable equivalent of the iminium and can regenerate it under the action of a Lewis acid. It can then react with different kind of nucleophiles. A last deprotection step gives access to the desired functionalized amine.
Scheme 2
This sequence can be achieved in good yields, and without technological difficulty. However, few asymmetric versions have been reported. The oxidation and alkylation of chiral carbamates have however been described. The yields of reaction were good but the diastereoselectivity observed was quite disappointing (d.e = 30% for piperidine and 60% for a pyrrolidine). We thought this result could be explained by the distance between the chiral center of the inductor and the reaction center. Our idea was then to use chiral heteroatom based structures as deactivating groups and chiral inductors.
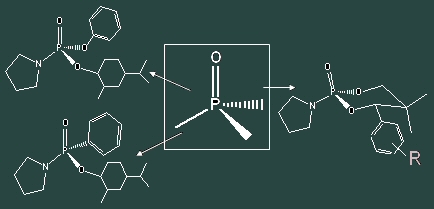
We chose phosphorus as our first choice of chiral heteroatom. It presented many advantages: it is widely use in all cases of asymmetric reactions (organometallic, deprotonation, ); it's an NMR probe, which is useful for our screening and it presents many tunable parameters (oxidation state of the phosphorus, number and nature of the substituents). We began with phosphoryl groups described in the literature as chiral NMR probes and amino acids resolution compounds. A family of chiral inductors was synthesized along with "open" forms. These five different groups were screened on a standard anodic oxidation- alkylation sequence using allyl TMS as a model nucleophile. We chose pyrrolidine as substrate. Results revealed one structure as the most interesting. This 60% e.d is comparable to the best one found in the literature, using a chiral carbamate. It is notable that in all cases yields of reaction; methoxylation as well as alkylation are very good to excellent, demonstrating once again the interest of this strategy. These results are presented in a first publication.
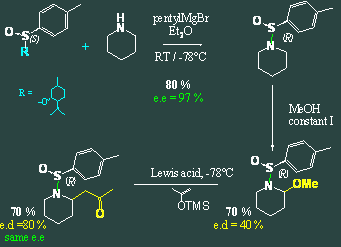
We then explored the limits of our best chiral inductor, expanding the scope to other amines. We tried and functionalized other cyclic secondary amines: piperidine, tetrahydroisoquinoline and morpholine. An acyclic secondary amine, diethylamine gave good results whereas primary amines could not be functionalized this way. Diverse nucleophiles have been screened too, demonstrating a difference between p and s nucleophiles. An other paper will summarize those results. In parallel, another chiral heteroatom was investigating in the lab: sulfur. I collaborated on this work, dealing with the first anodic oxidation of sulfonamides. It was possible to oxidize optically active sulfonamides and to alkylate them using silyl ether from acetone. The results obtained on piperidine, are really encouraging as diastereomeric excess of almost 90% could be obtained on this first creation of a chiral center. More details are given in the corresponding paper.
Scheme 4
|
||||||||||||||||||||
|
|
|
||||||||||||||||||||


Copyright(c) 2005. Webmaster Emma SIERECKI emma_sierecki@yahoo.fr